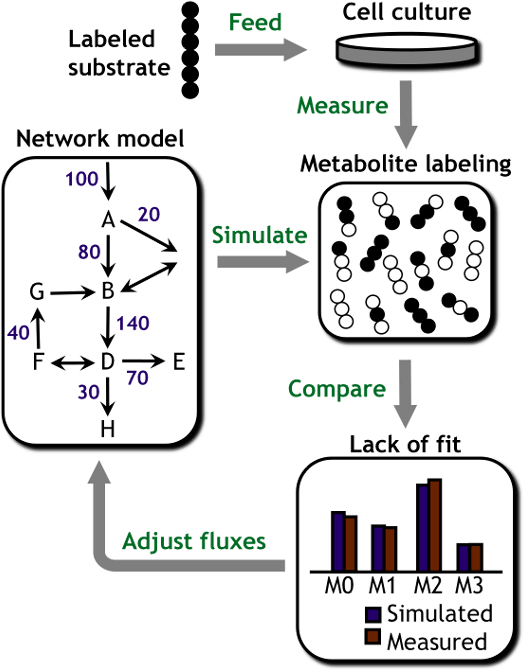
MFA studies are carried out by feeding cells an isotopically labeled substrate (e.g., 13C-labeled glucose) and subsequently measuring the patterns of isotope incorporation that emerge in downstream metabolites using mass spectrometry. A computational model of the intracellular metabolic network is used to determine pathway fluxes by integrating these isotope labeling data with additional measurements of extracellular nutrient uptake and product excretion rates. By systematically accounting for all extracellular carbon inputs and outputs and all major intracellular pathways, MFA can be used to reconstruct comprehensive flux maps depicting cell metabolism.
If the various biochemical pathways inside the cell are considered as highways, MFA is analogous to generating a traffic report describing the flow through these highways and how they change in response to a roadblock or detour. Comparison of flux maps obtained under varying experimental conditions or in the presence of targeted genetic manipulations provides a functional readout on the global impact these perturbations have on cell metabolism. Such information is essential to understanding how metabolic pathways are regulated in working cells—and how they become dysregulated in diseased cells. Furthermore, it enables us to pinpoint key metabolic nodes that can be manipulated in order to restore normal metabolic function to unhealthy cells (for biomedical applications) or to enhance production rates and yields in industrial host organisms (for biotechnology applications).
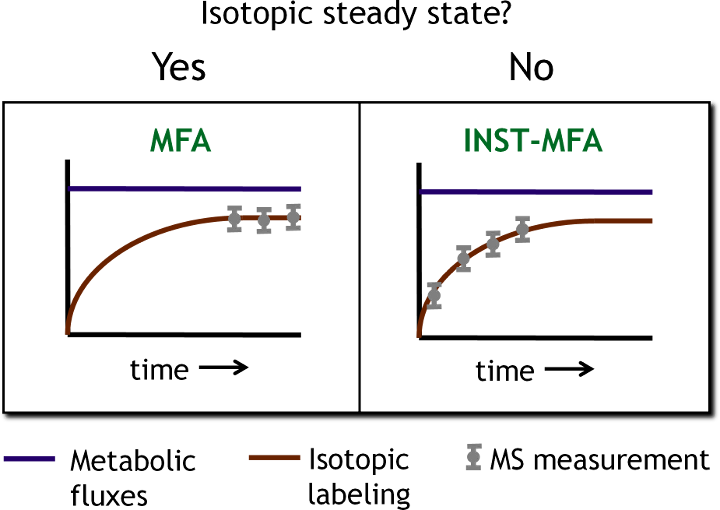 During his postdoctoral training, Dr. Young developed a novel MFA approach that uses transient isotope labeling experiments to determine metabolic fluxes, which is called isotopically nonstationary MFA (INST-MFA). Although the INST-MFA concept had been previously introduced by other groups, suitable computational tools for solving the large systems of ODEs that describe unsteady-state isotopomer balances were lacking. By applying an efficient decomposition of the isotopomer balance equations, an algorithm was developed that provides approximately 5000-fold computational speedup relative to previous approaches, thus making INST-MFA feasible for biologically relevant networks (
During his postdoctoral training, Dr. Young developed a novel MFA approach that uses transient isotope labeling experiments to determine metabolic fluxes, which is called isotopically nonstationary MFA (INST-MFA). Although the INST-MFA concept had been previously introduced by other groups, suitable computational tools for solving the large systems of ODEs that describe unsteady-state isotopomer balances were lacking. By applying an efficient decomposition of the isotopomer balance equations, an algorithm was developed that provides approximately 5000-fold computational speedup relative to previous approaches, thus making INST-MFA feasible for biologically relevant networks (